 Home
Home Tools
Tools Statistics
Statistics Advance
Advance Tutorial
Tutorial FAQ
FAQ Contact us
Contact usGene Ontology Enrichment Analysis Software Toolkit |
|
Home |
Tools |
Statistics |
Advance |
Tutorial |
FAQ |
Contact us |
GOEAST FAQIf you can not find answers to your questions here, please contact us. General GOEASTWhat is GOEAST?Gene Ontology Enrichment Analysis Software Toolkit (GOEAST) is a web-based software toolkit for fast identification of underlining biological relevance of high-throughput experimental results. GOEAST discovers statistically significantly enriched GO terms among the given gene list, and provides thorough, unbiased and visible results. What is GO?
Why should we use GOEAST?Various high-throughput experiments are becoming more and more important in this post-genomic era. However, high-throughput also indicates highly-complex and noise-generating. To easily understand the underlining biological relevance of a high-throughput experiment result, say a set of interesting genes/proteins, we have to find significantly enriched function of them and get rid of those might be selected by chance. Selecting the significantly enriched GO terms would be helpful a lot to understand our results. Who would be benefit from GOEAST?GOEAST is quite compatible for various high-throughput experiment results. Thus, users who use these techniques, such as microarray, 2-D gel electrophoresis of proteome, protein microarray, SAGE, MPSS, deep sequencing, would find GOEAST very helpful. GOEAST would save you a lot of time and funds! How does GOEAST utilize GO information?The information of GO is quit complicated and organized in a hierarchical manner. GOEAST parses the GO term relationship recursively in the latest GO ontology files. The results are stored in GOEAST's own database. GO information is updated regularly, currently at every weekend. How does GOEAST map gene identifiers to GO terms?GOEAST intents to map gene identifiers from the original data resources. Thus, commercial microarray probe identifiers are mapped to GO terms by their annotation files, and gene identifiers of various species are mapped to GO terms by gene-association files obtainted from Gene Ontology Website. GOEAST will follow this disciplin to map genes to GO terms, if it involved in future new tools. How often does GOEAST update?The functionality of GOEAST is relatively stable, and small changes would be made irregularly. However, GOEAST updates its own database for each tools, and different tools have different updating frequencies, based own the information they use. For example, Affymetrix® release their Gene Chip®s' annotation files every 4 months, so GOEAST tool for Affymetrix® platform updates every 4 months too. Tools of GOEASTWhy GOEAST is favourite to microarray results?There is no doubt that GOEAST pays more attention for microarray experiments than other high-throughput techniques. This is because microarray experiments are still the most widely used techniques of high-throughput techniques in almost every researching fields, such as a huge number of different gene expression microarray platforms, whole genome CGH microarrays, commercial and customized microarrays, exon microarrays, SNP/haplotype microarrays, microRNA microarrays... Another reason is that microarray experiment is often noisy and thus generates too many results to be checked one by one. Currently, GOEAST mainly supports gene expression microarrays. Which commercial platforms does GOEAST support?If the commercial microarray company releases their microarray annotation files regularly and makes them public available, GOEAST will try to make the support available as soon as possible. Currently, GOEAST microarray analysis tools support commercial microarray platforms from Affymetrix®, illumina® and Agilent®. See statistics for more details. You are welcome contact us to inform us the availability of other commercial microarray platforms. I cannot find support for the commercial microarray platform I used, what should I do?GOEAST cannot include supports for all commercial microarray platforms, but you are welcome to tell us where to find the annotation files for your commercial platforms., and we will include it in the future version. You can use the customized microarray platforms for your analysis if you know the GO annotation information of probes on your microarray. What is the meaning of "customized microarray platforms"?It is always needed to design and manufacture our own customized microarrays. This is usaully achieved by plotting cDNAs, PCR products or oligo DNA onto plane matrix by a robot. Anyone could design microarrays containing any set of probes suit for his/her own work, thus we often call them as "customized microarray platforms". How to use GOEAST tools for costomized microarray platforms?You have to prepare a GO annotation file for you customized microarray in order to use GOEAST analysis. Usually this is achieved by mapping your your probes to other gene identifers, such as UniProtKB, REFSEQ or Ensembl genes and use GO annotation of those genes as that of the probes on your customized microarrays. Notably, if long spotted PCR products instead of short synthetic oligo-probes were used, you could typically annotate you sequences by BLASTX searches against UniProtKB protein sequences at EBI database. The one who designed your customized microarrays would be know how to find GO annotation for probes on your microarray. Or you can ask a bioinformatician for help. Note that you only have to find directly associated GO terms for your probes/genes, and GOEAST will do the remaining jobs. The GO annotation file of your probes should be a plain-text file and of this format. What is the difference of the three kinds of illumina® gene identifiers?Unlike most other commerical microarray companies, illumina® uses three dependent identifiers to stand for genes on its microarrays. These three different gene identifiers, namely "target", "search_key" and "probeID", are very similar and can almost be used interchangeably. However, subtle differences do exist. First, for newly released illumina microarrays, the target identifer is obsolete and thus not recommended. Second, there are very few genes without corresponding search_key, thus we don't recommend using search_key as gene identifier. Third, for previously released illumina microarrays, probeID are 10-digit intergers with leading zeros. When opened by a spreedsheet such as Microsoft Excel, leading zeros of the probeID will disappear mysteriously! This will cause unnecessary problem if you upload your interesting genes by cutting-and-paste from a Microsoft Excel. If you intented to use probeID as gene identifier for old illumina microarrays, please be careful. GOEAST resultsHow to understand GOEAST graphical results?
The result graphs display enriched GOIDs and their hierarchical relationships in "biological process",
"cellular component" or "molecular function" GO categories. Boxes represent GO terms, labeled by its GOID,
term definition, p-value and detail information (see below). Significantly enriched GO terms are marked
yellow. The degree of color saturation of each node is positively correlated with the significance of
enrichment of the corresponding GO term. Non-significant GO terms within the hierarcical tree are either shown
as white boxes or drawn as points. Branches of the GO hierarchical tree without significant enriched GO terms
are not shown. Edges stand for connections between different GO terms. Red edges stand for relationship
between two enriched GO terms, black solid edges stand for relationship between enriched and unenriched terms,
black dashed edges stand for relationship between two unenriched GO terms. What are the meanings of every column in the GOEAST text results?All the 10 columns of GOEAST text format results are described below:
How does GOEAST calculate the Log odds-ratio for enrichment?Taken the definiton of q, m, k, t from the above section, the Log odds-ratio (LR) is defined as
How does GOEAST calculate the p-value for enrichment by default? Why?By default, the p-value of GOID enrichment is calculated as the hypergeometric probability to get so many probes/probesets/genes for a GO term, under the null hyperthesis that they were picked out randomly from the microarray/genome. To be specific, the p-value can be calculated as
Why GOEAST recommend doing multi-test adjustment for the p-value?P-value is used for control the type I error rate in one statistical test. But when GOEAST identifies significant enriched GO terms, the same type of statistical tests was carried out many times. Under such circumstance, controling the false discovery rate (FDR) of the whole results would become important. GOEAST provides FDR value by default, but such multi-test adjustment could be disabled as required. What kind of method does GOEAST use to adjust the p-value by default? Why?There are several methods to adjusting the raw p-value to FDR for different types of data. Because individual tests in GOEAST would be positively related, especially for GO terms on same hierarchical trees, we use Benjamini & Yekutieli (2001) method to calculate the FDR value by default. Other method to circumvent the multi-test problem supported by GOEAST includes Hochberg FDR, Bonferroni, Hochberg, Hommel method. I cannot see the figures of the graphical output, what could I do?The default output figures are in Adobe PDF format and could be viewed using Adobe Reader. Click here to download Adobe Reader. The output figures in SVG format could be viewed in browser after installing Acrobat SVGViewer plug-in. Click here to download Acrobat SVGViewer. Can GOEAST make more annotation for genes of its analysis results?For commerical microarray platforms, GOEAST only displays several important annotation for every genes, due to space limitation. For customized microarray platforms, you could supply (optional) probe/gene annotations, which will be displayed later. I found an obsolete GO term in my GOEAST result, why?Because the update of the GO is very frequently (every 30 minutes), but microarray annotation files update much less frequently. Therefore, GO terms in the microarray annotation might become obsolete in GO annotation. GOEAST AdvanceWhat is the Adrian Alexa's improved weighted scoring algorithm? What's it advantages and limitations?In graph algrithm, the GO hierarchy can be represented by a DAG (Directed Acyclic Graph), whose nodes (vertices) represent for GO terms and edges for parents-children relations. Thus, as mentioned above, closely related GO terms often positively correlate in GO enrichment analysis. The Adrian Alexa's algorithm is an improved method to de-correlate these correlations in the GO DAG. Generally speaking, it is achieved by down-weighting genes in less significant neighbors of all GO terms in a botton-up manner. Detail description of the algorithm can be found here. Adrian Alexa's algorithm brings strong power at controlling the false discoveries while not reducing the sensitivity of GO enrichment analysis significantly. GOEAST implements Adrian Alexa's improved weighted scoring algorithm by using R package "topGO" from Bioconductor. However, this algorithm is quit expensive both in time and spacing, and GOEAST won't use this algorithm by default. What is the difference between Batch-Genes and normal GOEAST tools?Batch-Genes tool uses different kind of statistical backgrounds compared to normal GOEAST tools. To be specific, Batch-Genes use per-species backgrounds whereas other tools use per-array backgrounds. Thus, it is more suitable for non-microarray based experiments. Both Batch-Genes and GOEAST use same statistical method to calculate p-values or FDRs. I have transformed my probes into gene accessions for my microarray, could I use Batch-Genes instead?Yes, you could, but we recommend you to stick with the normal GOEAST tool. Using Batch-Genes might involve in unnecessary system bias, especially for those non-genome microarray platforms. What gene ID formats dose GOEAST support in Batch-Genes tool?In Batch-Genes tool, different species requires different gene ID formats. As the GO annotation is achieved by different organizations for different species, thus GOEAST uses different gene ID formats for different species. Nevertheless, some databases support GO annotation for multiple species, such as UniProtKB, and thus could be used for several species. Why Batch-Genes doesn't include the species I am studing with?Batch-Genes now supports most model organisms used in biological reserches. More species would be included if the interesting on it were higher and studies became more. How can I understand the result figures of Multi-GOEAST?Multi-GOEAST result figures can be considered as the combination of several normal GOEAST result figures. GO nodes existing in any individual uploaded files are draw as boxes. The background color for all nodes are the combination color of each uploaded files you set. Edges are drawn the same way as normal GOEAST tools. Will Multi-GOEAST support analysis of >3 inputs in the future?It is not because we can't but we wouldn't like to support >3 inputs for Multi-GOEAST. Multi-GOEAST is designed to make it convenient to compare multiple GOEAST results. Using the color combination strategy, users could easily find out common or specific GO terms enriched in different results. As the combinded color is unimaginable if we combine more than 3 colors, we don't support >3 inputs. Performance & miscellaneous itemsHow fast does GOEAST work?It depends. The calculation time on different microarray platforms and different species will be different. Closely related gene sets will take less time than un-related gene sets. Typically a query of nearly 1,000 genes would take from 2 minutes to a maximum 45 minutes, but it might be slower if our server is heavily loaded. If you haven't got your result or the finishing email after an hour, please try again later. At last, if the Adrian Alexa's algorithm is selected in an analysis, it will require an extra time of up to 15 minutes. Which web browsers does GOEAST support?All GOEAST web pages follow the W3C recommended XHTML1.0 DTD standard, which are compatible with most modern web browsers, including Internet Explorer, Firefox, Mozilla, Safari, Konqueror, Opera, Netscape, America Online, et.al. |



| Institute of Genetics and Developmental Biology, Chinese Academy of Sciences ©Version 1.30 Copy Right 2007-2016 GOEAST org. Beijing ICP No. 09063187 |
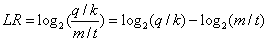 . The bigger LR is, the higher the relative abundance of this GO term is compared to random situation.
. The bigger LR is, the higher the relative abundance of this GO term is compared to random situation.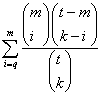 . The smaller the p-value is, the more significant the GO term is enriched in your dataset. Other statistical test is also supported to calculate p-value in advanced paramter setteings, e.g. Fisher exact test and Χ2 test. However, Fisher exact test is statistically equivalent to hypergeometric test, which can be calculated much faster in R. Χ2 test is only suitable for GO terms containing very lot genes. Thus, GOEAST uses hypergeometric test by default.
. The smaller the p-value is, the more significant the GO term is enriched in your dataset. Other statistical test is also supported to calculate p-value in advanced paramter setteings, e.g. Fisher exact test and Χ2 test. However, Fisher exact test is statistically equivalent to hypergeometric test, which can be calculated much faster in R. Χ2 test is only suitable for GO terms containing very lot genes. Thus, GOEAST uses hypergeometric test by default.